Inhaltsverzeichnis
Lysosomen sind kugelförmige Zellorganellen, die auch als Magen der Zelle bezeichnet werden. Warum man sie so nennt und wie sie aufgebaut sind, behandelt dieser Artikel.
Inhaltsverzeichnis
Lysosomen – Definition
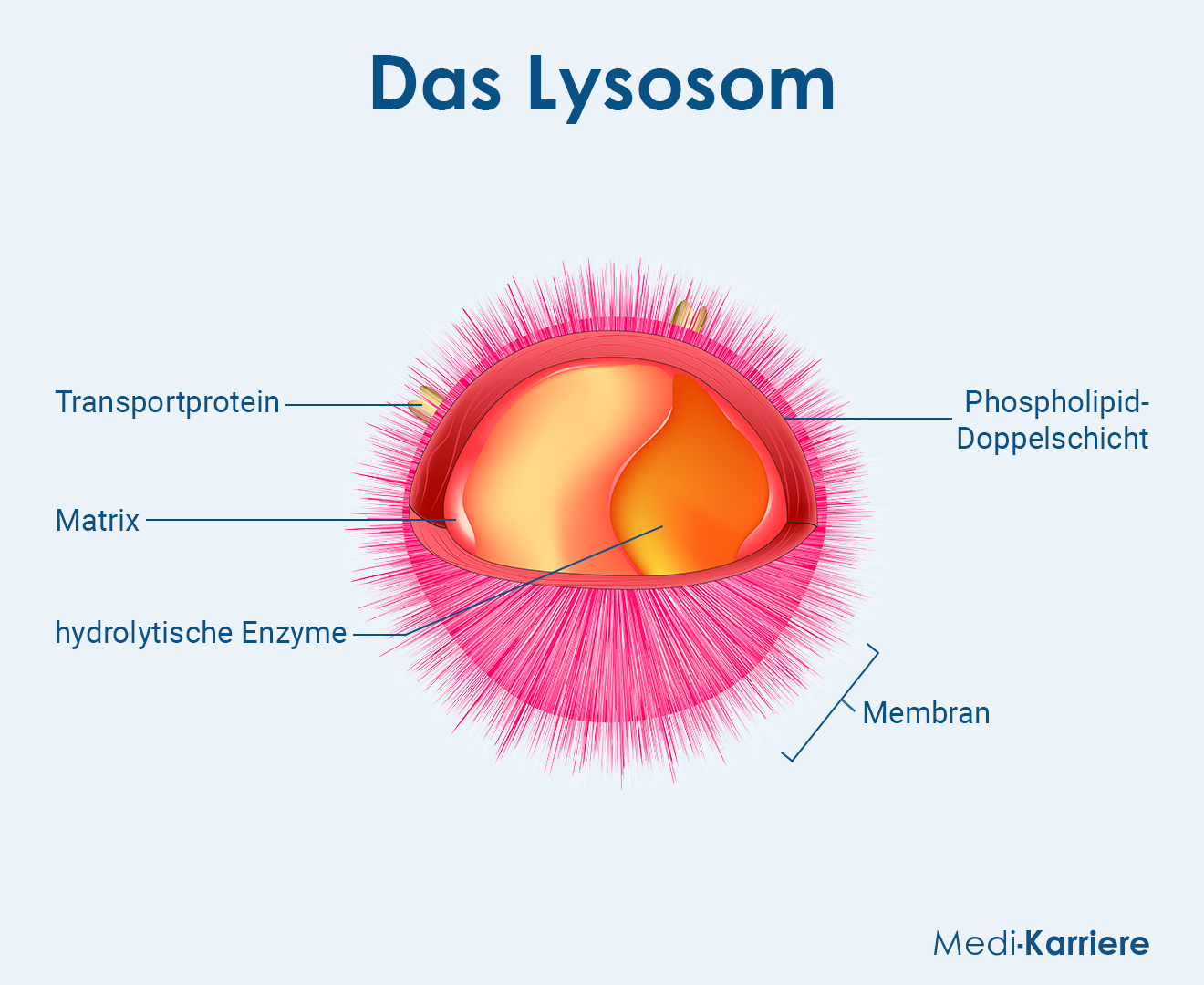
Unter Lysosomen versteht man von einer Membran umschlossene Zellorganelle, die in eukaryotischen Zellen vorkommen und in etwa wie kugelförmige Bläschen aussehen. Sie beinhalten hydrolytische Enzyme und Phosphatasen, mithilfe derer sie Fremdstoffe oder auch körpereigene Stoffe abbauen können.
Lysosomen – Aufbau
Im Inneren der Lysosomen befinden sich zahlreiche hydrolysierende Enzyme wie Nukleasen, Proteasen und Lipasen. Sie dienen der intrazellulären Verdauung von nicht mehr gebrauchten Materialien und erreichen nur im sauren Milieu eine hohe Aktivität. Dieses saure Milieu ist im Inneren eines Lysosoms gegeben, da dort der pH-Wert unter fünf liegt. Die Notwendigkeit eines sauren Milieus schützt andere Zellorganellen für den Fall, dass Enzyme aus den geschädigten Lysosomen in das Zytosol gelangen. Dort herrscht ein alkalischeres Milieu, sodass die Enzyme nicht aktiv sind und keine wichtigen zellulären Bestandteile abbauen.
Der saure pH-Wert in den Lysosomen wird durch eine spezielle ATPase aufrecht erhalten. Diese transportiert unter der Hydrolyse von einem ATP-Molekül zwei Protonen in das Lysosom. Des Weiteren zeichnet sich die Innenseite der Lysosomenmembran durch speziell glykosylierte Proteine aus, damit die Enzyme im Inneren nicht auch die Zellmembran angreifen und beschädigen.
Lysosomen – Funktion
Die Hauptaufgabe von Lysosomen ist der Abbau von Fremdstoffen oder auch körpereigenen Stoffen. Dafür werden die lysosomalen Enzyme benötigt. Die Synthese dieser findet im endoplasmatischen Retikulum statt, woraufhin sie in dessen Lumen abgegeben werden. Dann werden sie in Transportvesikel verpackt und gelangen zu den Lysosomen.
Auf dem Weg dorthin kommen sie am Golgi-Apparat vorbei. Dort wird entschieden, ob das Enzym aus der Zelle herausgeschleust oder zu anderen Zellorganellen weitergegeben werden soll. Um sicher zu stellen, dass die lysosomalen Enzyme in die richtigen Vesikel geladen werden und auch wirklich in den Lysosomen ankommen, werden sie mit einem bestimmten Marker gekennzeichnet. Bei den Hydrolasen findet die Markierung mittels des Kohlenhydrats Mannose statt, welches im Golgi-Apparat phosphoryliert wird. Diese Modifikation wird von zwei Enzymen katalysiert. Eine Phosphotransferase erkennt, dass es sich um ein lysosomales Enzym handelt und heftet N-Acetylglucosamin-1-phosphat an ein oder zwei Mannosereste. Das zweite Enzym schneidet dann den N-Acetylglucosamin-Rest ab, womit die Markierung durchgeführt ist. Der Marker wird dann anschließend von speziellen Rezeptorproteinen erkannt, die für die Beladung in die korrekten Vesikel sorgen.
Lysosomen – Lysosomale Speicherkrankheiten
Lysosomale Speicherkrankheiten entstehen dann, wenn ein Defekt in der Phosphotransferase vorliegt und somit keine Markierung stattfinden kann. Die lysosomalen Enzymen werden in diesem Fall nicht sortiert und gelangen unkontrolliert in die extrazelluläre Matrix. Andere lysosomale Speicherkrankheiten werden wiederum dadurch ausgelöst, dass die lysosomalen Hydrolasen selbst defekt sind. Als Folge vermehrt sich nicht aufgebautes Material in den Lysosomen und kann für schwere Krankheitssymptome sorgen.
Der Begriff lysosomale Speicherkrankheiten ist als Sammelbegriff zu verstehen, mit dem zur Zeit 45 Krankheiten zusammengefasst werden.
Häufigkeit von lysosomalen Speicherkrankheiten
Lysosomale Speicherkrankheiten sind insgesamt relativ selten und entstehen in der Regel durch Mutationen in Genen, die für die betroffenen Enzyme codieren. Im Durchschnitt ist in etwa eines von 8.000 Neugeborenen von dieser Erkrankung betroffen.
Die nicht abgebauten Materialien sammeln sich zuerst in der Zelle und im weiteren Verlauf auch im Extrazellulärraum an. Besonders betroffen sind Zellen von Leber, Knochen, Nervensystem, Haut und Milz. Je nach den Stoffen, die am meisten akkumulieren, teilt man die lysosomalen Speicherkrankheiten ein. Dadurch ergeben sich folgende Typen:
- Lipidosen
- Mukopolysaccharidosen
- Oligosaccharidosen
- Mukolipidosen
Zu den am häufigsten vorkommenden lysosomalen Speicherkrankheiten zählen der Morbus Gaucher, der Morbus Fabry und Morbus Pompe. Dem Morbus Gaucher, der am häufigsten vorkommt, liegt eine verringerte Aktivität der Glucocerebrosidase vor. Dies führt zu einer Anreicherung von Glucosylceramid in Zellen des retikuloendothelialen Systems. Als Folge davon kommt es zu Symptomen wie der Vergrößerung von Leber und Milz, Störungen des Skelettsystems, hämatologische Veränderungen, in seltenen Fällen Neurodegeneration, epileptische Anfälle und Lungenbefall. Bei Säuglingen können außerdem Fütterstörungen auftreten. Die Erkrankung kann durch die Bestimmung der Aktivität der Glucocerebrosidase in Leukozyten oder Haut–Fibroblasten diagnostiziert werden.

- Lüllmann-Rauch, Renate: Taschenlehrbuch Histologie, Thieme (Stuttgart: 6. Auflage, 2019)
- Aumüller, Gerhard et al.: Duale Reihe Anatomie, Thieme (Stuttgart: 5. Auflage, 2020)



